e-DrugMod
Molecular modeling for drug design


AMMOS2: Automatic Molecular Mechanics Optimization for in silico Screening
AMMOS2 (Automatic Molecular Mechanics Optimization for in silico Screening) employs an automatic procedure for energy minimization of protein-ligands complexes.
 Run a job
Run a job Job results
Job results
AMMOS (Automatic Molecular Mechanics Optimization for in silico Screening)[Pencheva 2008] is a software platform that employs an automatic procedure for energy minimization of protein-ligands complexes via the website AMMOS2 and of small chemical compounds via the website AMMOS. AMMOS offers valuable solutions to assist structure-based in silico screening experiments or ligand-based projects. It makes use of molecular mechanics concepts and is based on the program AMMP [Harrison 1997, Weber 1998], available under GNU license. AMMOS has been developed by the QSAR and Molecular Modelling Dep., IBBE, BAS and the MTi lab, INSERM U973 – University Paris Diderot. Please, cite:Pencheva T, Lagorce D, Pajeva I, Villoutreix B, Miteva MA., AMMOS: Automated Molecular Mechanics Optimization Tool for in silico Screening, BMC Bioinformatics, 2008, 9:438Jereva D, Pencheva T, Lagorce D, Desvillechabrol D, Pajeva I, Miteva MA. Post-docking optimization of protein-ligand interactions involving water molecules. Asian J Physics. 2014; 23: 745-56 You may direct questions related to AMMOS to:Maria A. MitevaMTi, INSERM U973 - University Paris DiderotE-mail: maria.mitev@inserm.fr
AMMOS2 performs energy minimization of protein-ligand complexes and can be applied on a large number of experimental or modeled protein-ligand complex structures, i.e. pre-generated via a user-chosen docking programs or e.g. via the MTiOpenScreen web server. The molecular mechanics minimization of AMMOS2 is based on the AMMP force field sp4 [Bagossi 1999] developed by use of the UFF potential set [ Rappe et al. 1992]. Intermolecular interactions include Electrostatic and van der Waals interactions. The minimization is realized by performing 2×500 conjugate gradient iterations. AMMOS2 requires as input a protein file in pdb format and docked small ligands in mol2 format. The translation of small molecule and protein files format in a specific ammp format is executed by the PREAMMP program, which is a part of the AMMP package and the AMMOS2 web service. protein flexibility AMMOS2 allows optimization of proteins - small organic molecules interaction at five levels of flexibility of the protein receptor, while ligands are always flexible:Allowed to be flexible:
protein flexibility AMMOS2 allows optimization of proteins - small organic molecules interaction at five levels of flexibility of the protein receptor, while ligands are always flexible:Allowed to be flexible:
- Case 1: entire protein
- Case 2: protein sidechains
- Case 3: all protein atoms within a sphere of up to 8 Å of ligand atoms
- Case 4: protein sidechain atoms within a sphere of up to 8 Å of ligand atoms
- Case 5: the entire protein is rigid
Ligand atomsThe ligand atoms supported by AMMOS2 are : H, C, B, N, O, P, S, Cl, Br, I, F, Fe, Na, K, Mg, Ca, Zn.water moleculesThe AMMOS2 version allows to include explicit water molecules in the protein-ligand complex. Water molecules that can be accommodated in protein binding site are of key importance for drug design. AMMOS2 allows an inclusion of explicit water molecules mediating protein-ligand interactions or water molecules around the binding site for the molecular mechanics optimization of the system.metal ionsAMMOS2 takes into account the presence of individual metal ions, which are apart of co-factors. The ions of Na, K, Mg, Ca, Zn and Fe are taken into account in the AMMOS2 molecular mechanics optimization and are allowed to move in flexibility cases 1 to 4.
The entire procedure of AMMOS2 is shown in the following scheme:
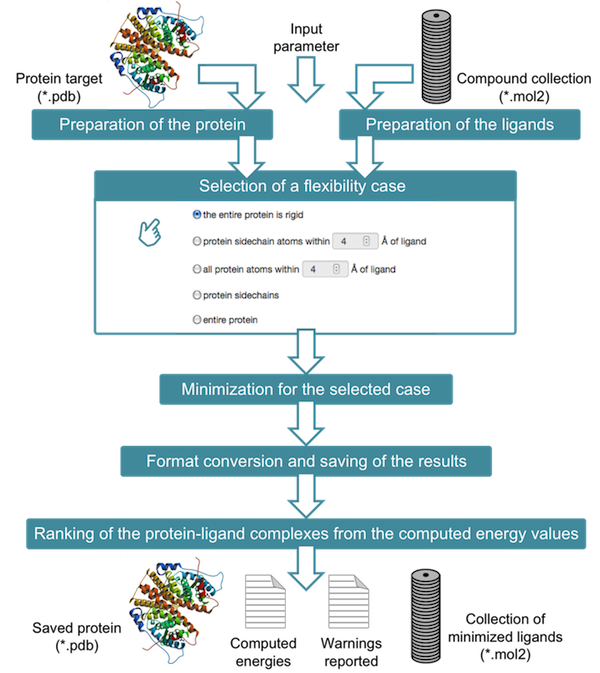
AMMOS2 requires two input files: the protein in pdb format and a compound collection containing the pre-docked ligands in mol2 format. Before to proceed to AMMOS2 use, please consider following requirements:Protein and ligand namesPlease, ensure that the protein and ligand file names do not contain any spaces.The protein PDB file name should finish by .pdbThe ligand MOL2 file name should finish by .mol2The field of charges in the mol2 file should be provided or assigned to 0.0000.How to prepare the proteinAMMOS2 requires a protein in pdb format with no more than 1000 residues. It is expected the protein to be properly protonated by the user. The hydrogen atom names should correspond to atom names assigned by CHIMERA.The format of HETATM field of the input PDB should be like:"HETATM 4976 O HOH A2500 -33.154 84.346 62.465 1.00 68.42 O""HETATM 3234 CA CA A 224 37.739 25.336 34.972 1.00 0.00 Ca" For FE2 and FE3, please use residue name FE2 or FE3. and not like:"HETATM15147 O HOH A2058 -30.531 68.972 12.744 1.00 34.99 O""HETATM 3234 CA CA3 A 224 37.739 25.336 34.972 1.00 0.00 Ca"How to prepare the ligandsAMMOS2 requires a compound collection containing the pre-docked ligands in mol2 format. A collection of up to 300 ligands is acceptable for the Cases 1, 2 and 5, while for the Cases 3 and 4, collections of up to 2000 ligands might be introduced. For all Cases, the maximum number of atoms in the ligand should not exceed the number of 300, including hydrogens. Ligand hydrogens can be provided by the user or added by AMMOS2.Radius of the binding siteThe user-defined radius of a sphere around the ligand atoms for Cases 3 and 4 should be between 4 and 8 Å.How to run
- Choose a protein in pdb format using button “Choose file” in Block “1-Protein”
- Allow a part of protein to be flexible selecting a flexibility case in Block “1-Protein”
- Choose a compound collection containing the pre-docked ligands in mol2 format using button “Choose file” in Block “2-Ligands”
- Keep the ligand protonation choosing/not choosing “Yes” in Block “2-Ligands”
- Choose an option to be/or not informed by email when the job is finished in Block “3-Submit”
- Submit your task using button “Submit” in Block “3-Submit”
- the entire protein is rigid
- protein sidechain atoms within a sphere of up to 8 Å of ligand
- all atoms within a sphere of up to 8 Å of ligand
- protein sidechains
- entire protein.
Your results will be kept during 2 weeks after the job terminationPlease, keep your jobID.After the job submission the page will be refreshed automatically every 30 seconds. After the job termination you can download the results as a tar archive. The tar file contains:
- "protein.pdb" containing your input protein file
- "energy.txt" containing the energies before and after minimization
- "minimized_ligands.mol2" containing all renumbered minimized ligand structures ranked by the predicted binding energy noted in the energy.txt as "AFTER_EXTERNAL"
- Ligand_directory containing:
- "ligand_minimized.mol2" containing the minimized ligand structure
- "plip_prepared_output.pdb" containing the minimized protein and minimized ligand for cases 1-4 and the input protein and minimized ligand for case 5
- "PLIP.png" and "report.txt" containing a figure and information on the interactions in the minimized protein-ligand complex
- log and warning files
Visualization:The binding energies for the top 100 ranked ligands are displayed in a table. By clicking on the ligand name the corresponding complex will be loaded in the viewer on the right side of the page. In this viewer, the input protein is shown in grey cartoon and the input water molecules are shown in grey. The minimized ligand is shown in atom type sticks. The minimized waters are shown in dark blue balls and sticks. The minimized protein is shown in orange cartoon for Case1 and in grey atom type for Cases 2-4.By clicking on the ligand name in the table the corresponding minimized protein-ligand interactions generated by PLIP will be loaded in the viewer on the left side of the page.
- an automatic procedure for energy minimization of protein-ligands complexes at five levels of flexibility of the protein receptor, while ligands are always flexible
- allowing inclusion of explicit water molecules in the protein-ligand complex
- allowing inclusion of individual metal ions, which are apart of co-factors
- visualization in 3D of protein-ligand contacts after the minimization via PLIP (Salentin,S. et al. PLIP: fully automated protein-ligand interaction profiler. Nucl. Acids Res. (1 July 2015) 43 (W1): W443-W447. doi: 10.1093/nar/gkv315) for the top 100 ranked ligands scored by the predicted interaction energy between the protein and the ligand
- visualization in 3D of the entire minimized protein-ligand complex for the top 100 ranked ligands scored by the predicted interaction energy between the protein and the ligand
- download of the minimized ligand structures in MOL2
- download of the minimized complex structures in PDB
- Protein residues are limited to 1000
- Protein should be properly protonated by the user
- AMMOS2 does not include co-factors in the minimization except individual metal ions. If present in the input PDB file, they will be automatically deleted
- AMMOS2 cannot treat covalently bound ligands
- The number of ligands is limited to 1000 for Cases 1, 2 and 5
- The number of ligands is limited to 5000 for Cases 3 and 4
- Ligands should not exceed 300 atoms
Ammos2 web server has been rigorously benchmarked for the performance of our implementation of the Ammos software. Ammos2 molecular mechanics minimization performance has been validated against 21 diverse and extensively checked protein-ligand complexes from the PDB (CCDC/Astex Test Set). We selected several classes of important protein targets (nuclear receptors, kinases, serine proteases, ribonucleases among others) with available high quality crystal structures manually selected from the Astex test dataset. The ability of Ammos2 to improve the protein-ligand interaction predictions was validated after molecular docking of co-crystallized ligands by using AutoDock4. The molecular mechanics optimization of docked protein-ligand complexes was performed by Ammos2 for five different cases of protein flexibility, in presence or absence of explicit water molecules located in the binding pocket or in the whole protein. The impact of considering water molecules during the minimization on the ligand binding modes and binding energies was assessed. Two examples are shown here for CAMP-dependent protein kinase (PDB ID: 1yds, 1ydr, 1ydt) and trypsin (PDB ID: 1mtw, 1mts, and 1ql7). Including protein flexibility during the minimization with Ammos2 optimized the interactions in the complexes, resulting in more favorable interaction energies. Adding several water molecules in the binding site also improved the protein-ligand interaction energies and optimized the positions of waters mediating the ligand-protein interactions.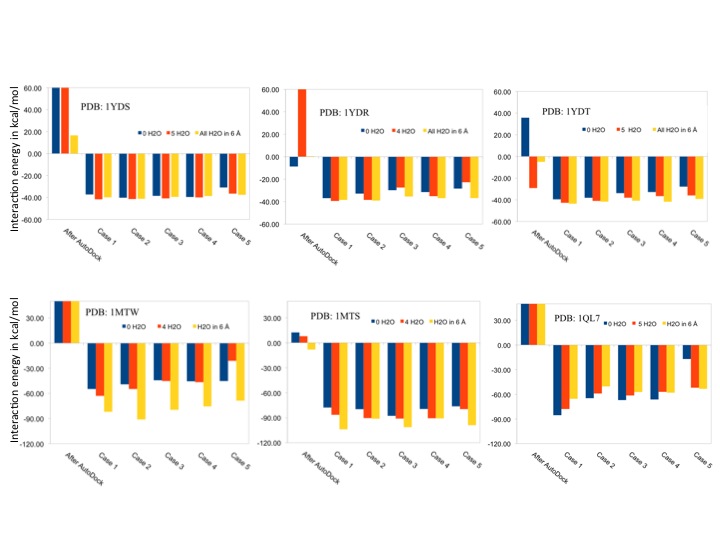 Figure 1. Binding energies of protein-ligand complexes minimized by Ammos2 in absence of water (0 H2O), or in presence of selected 4 or 5 water molecules in the binding site, on in presence of all X-ray water molecules within 6 Å of the ligand.
Figure 1. Binding energies of protein-ligand complexes minimized by Ammos2 in absence of water (0 H2O), or in presence of selected 4 or 5 water molecules in the binding site, on in presence of all X-ray water molecules within 6 Å of the ligand.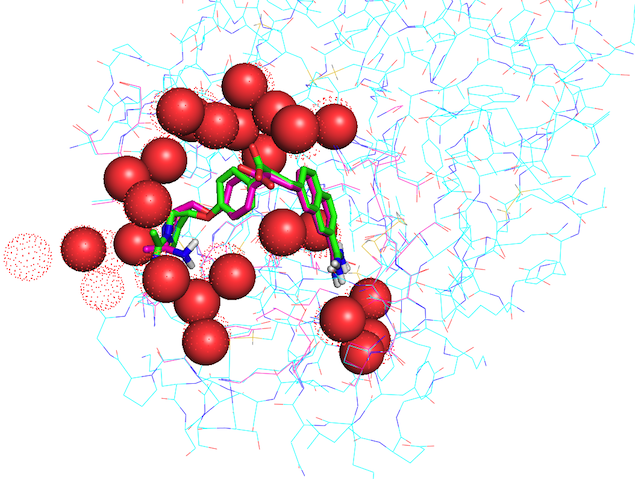 Figure 2. The X-ray structure of trypsin (PDB ID 1MTS) and its co-crystallized ligand are shown in cyan and green, respectively; water molecules are shown as dots. The protein and ligand minimized by Ammos2 Case 3 with present waters (shown as red spheres) within 6 Å of the ligand are shown in pink.
Figure 2. The X-ray structure of trypsin (PDB ID 1MTS) and its co-crystallized ligand are shown in cyan and green, respectively; water molecules are shown as dots. The protein and ligand minimized by Ammos2 Case 3 with present waters (shown as red spheres) within 6 Å of the ligand are shown in pink.
Bagossi P., G. Zahuczky, J. Tözsér, I. Weber, R. Harrison. Improved Parameters for Generating Partial Charges: Correlation with Observed Dipole Moments, Journal of Molecular Modeling, 1999, 5, 143-152Harrison R., C. Reed, I. Weber. Analysis of Comparative Modeling Predictions for CASP2 Targets 1, 3, 9, and 17. Proteins: Structure, Function, and Genetics, 1997, Suppl. 1, 68-73Jereva D., T. Pencheva, D. Lagorce, D. Desvillechabrol, I. Pajeva, M. Miteva, Post-docking Optimization of Protein-Ligand Interactions Involving Water Molecules, Asian Journal of Physics, 2014, 23, 745-756Pencheva T., D. Lagorce, I. Pajeva, Br. Villoutreix, M. Miteva, AMMOS: Automated Molecular Mechanics Optimization Tool for in silico Screening, BMC Bioinformatics, 2008, 9:438Pencheva T., D. Lagorce, I. Pajeva, B. O. Villoutreix, M. A. Miteva, AMMOS Software: Method and Application, Methods Mol Biol, Computational Drug Discovery and Design, R. Baron (Ed.), Humana Press, 2012, 819, 127-141Pencheva T., O. S. Soumana, I. Pajeva, M. A. Miteva, Post-docking Virtual Screening of Diverse Binding Pockets: Comparative Study Using DOCK, AMMOS, X-Score and FRED Scoring Functions, Eur J Med Chem, 2010, 45, 2622-2628Rappé AK, Casewit CJ, Colwell KS, Goddard WA, III, Skiff WM. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc, 1992, 114, 10024–10035Weber I., R. Harrison. Molecular Mechanics Calculations on Protein–Ligand Complexes. Perspectives in Drug Discovery and Design, 1998, 9/10/11, 115-127
The data associated with any job will be kept for a maximum time period of 7 days.All data that you enter or get from our Website are being stored on our servers. Data are not shared with other users or disclosed in any manner according to the standard protection rules: each job has an unique identifier coded upon 15 characters, logged sessions are protected by passwords. The RPBS team does not perform any analysis on the processed data.